
NF-κB是一种关键的细胞核转录因子,在细胞对多种刺激做出反应时发挥着核心作用。那么关于NF-κB信号通路有什么课题设计思路呢?今天我们一起来探讨一下。
接下来我们将分享一篇发表于中科院1区期刊Advanced Science,影响因子为14.3的文献,希望能给大家带来不一样的灵感。
一、文章信息

二、研究背景
①胰腺肿瘤发生发展涉及多种基因变异,包括KRAS、TP53、SMAD4和CDKN2A等。其中,90%左右的胰腺导管腺癌(PDAC)患者存在KRAS突变;
②NF-κB信号在近70%的胰腺癌标本中被组成性激活,并在PDAC的发展、进展和耐药性中起着至关重要的作用;
③NSD2是一种组蛋白甲基转移酶,可催化组蛋白H3在赖氨酸36(H3K36me2)位点的二甲基化,H3K36me2是一种与活性基因转录相关的允许标记;
④许多研究发现NSD2促进细胞增殖、迁移、侵袭和上皮-间质转化(EMT),证明其起着至关重要的促癌作用。
三、技术路线
 、
、
四、研究结果
1.NSD2过表达抑制炎症和/或KRAS突变诱导的胰管化生
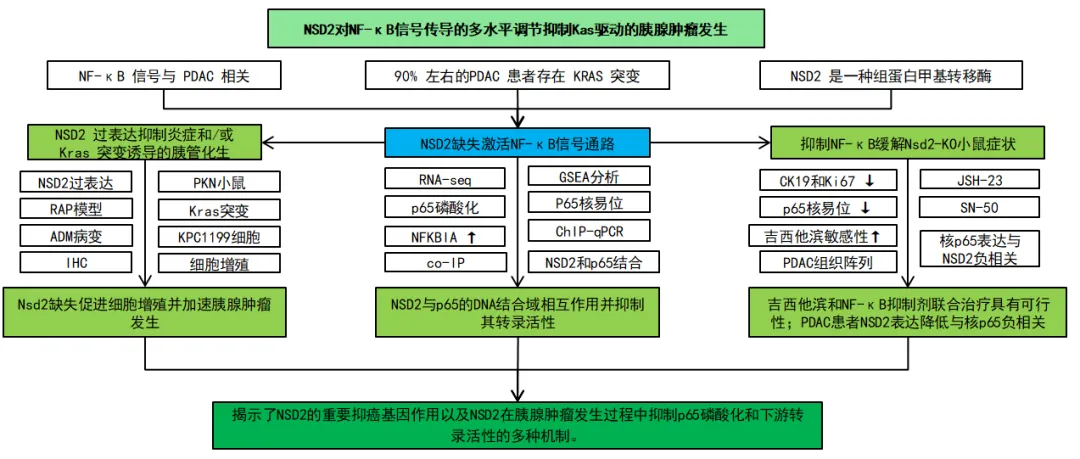
为了探究NSD2对炎症和KRAS突变的影响,研究团队把胰腺特异性NSD2过表达以及致癌KRAS突变小鼠称为PKN小鼠,在图1B~C中,WB和IHC验证了NSD2在PKN胰腺中过表达。而在使用caerulein诱导胰腺炎恢复RAP模型后,如图1E~F所示,PKN小鼠胰腺中腺泡到导管化生(ADM)病变在第7天和第18天都少得多,这表明NSD2过表达显著抑制了炎症诱导的ADM。图1G~H显示,PKN小鼠的ADM、PanIN和PDAC病变的发展显著减少,图1I也表明CK19+和Ki67+信号在PKN病变中显著降低。此外,在图1J~K中,PKN小鼠的胰腺腺泡外植体在响应TGFα的情况下,分化为导管样结构的程度较低。这些发现都共同表明NSD2过表达加强了腺泡稳态并抑制了炎症和KRASG12D诱导的胰管化生。
2.NSD2缺失促进KRAS诱导的导管化生
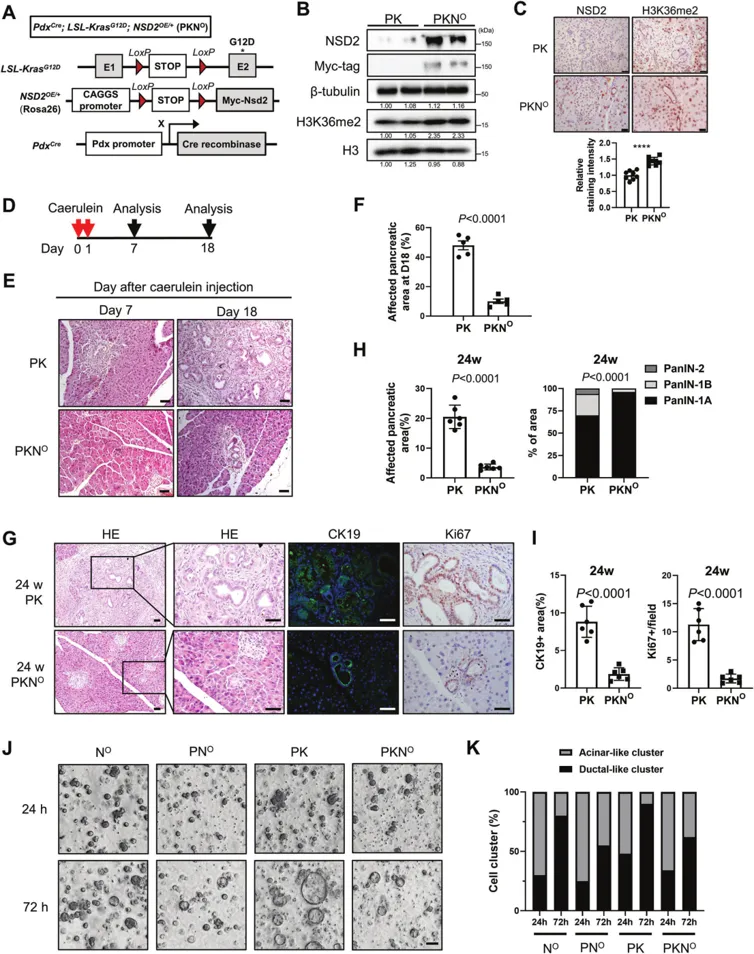
为了验证以上结果,团队进一步探索了NSD2缺失的影响,如图2A~C所示,通过WB、IHC和RT-qPCR实验,发现NSD2确实被彻底消融后,PKNf/f小鼠胰腺中的H3K36me2水平没有变化。而在图2D~G中,致癌KRAS突变的PK小鼠在12周龄时出现罕见的ADM病变,并在24周龄时逐渐进展为低级别PanIN。图2H显示,来源于PdxCre;NSD2f/f小鼠的胰腺腺泡外植体导管样结构比对照小鼠(NSD2f/f小鼠)分化程度更高。总之,NSD2丢失与致癌KRAS协同作用,共同促进ADM转变和向PanIN和PDAC的进展。
为了进一步分析NSD2缺失的影响,在小鼠胰腺癌细胞系KPC1199中沉默NSD2,图2I~J中可以看到,WB结果显示NSD2减少,但H3K36me2没有减少,与增加的Ki67+比例一致,CCK8证实了NSD2丢失诱导的细胞增殖增加。这些结果表明NSD2缺失促进细胞增殖并加速胰腺肿瘤发生。
3.NSD2缺失激活NF-κB信号通路
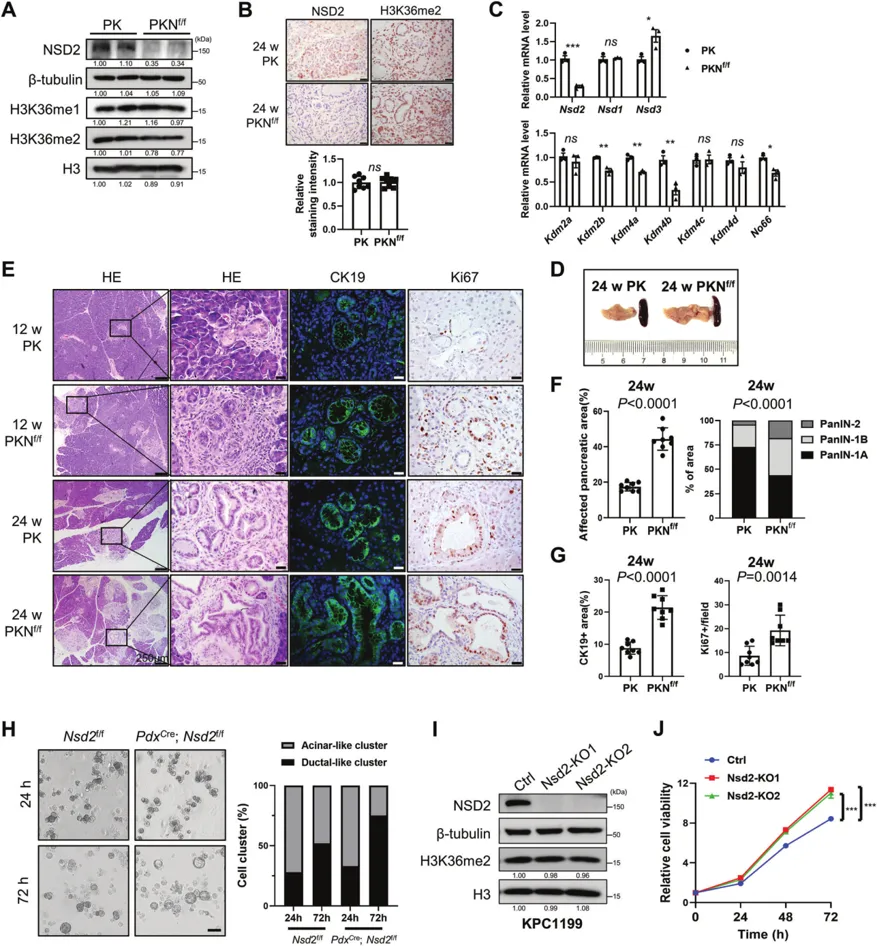
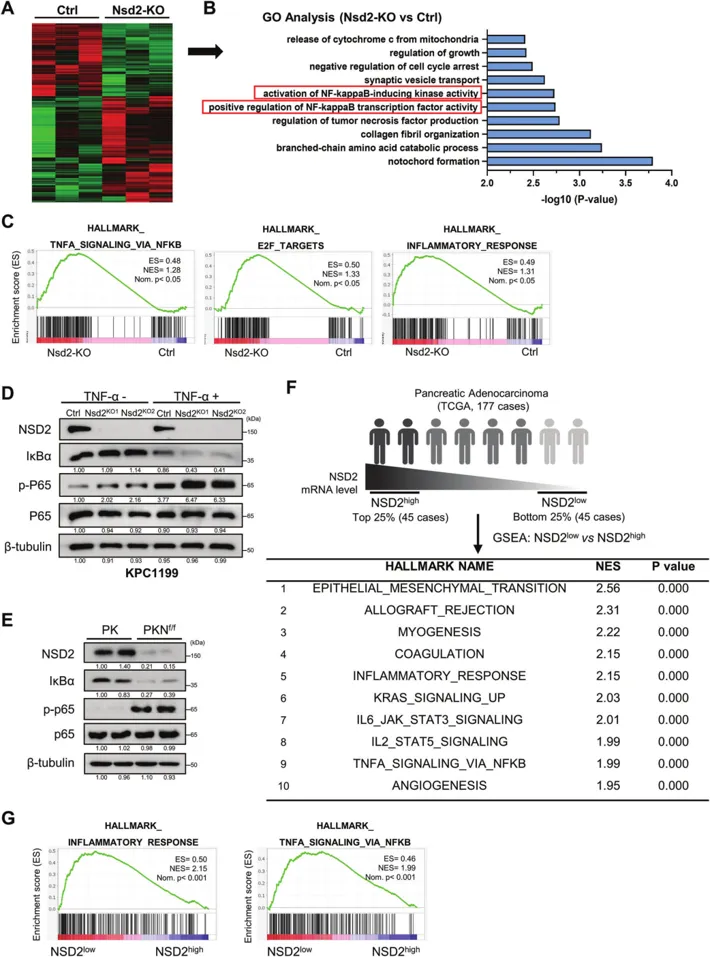
紧接着,他们针对NSD2缺失与NF-κB信号通路激活之间的联系展开研究,图3B中的RNA-seq分析结果,显示了NSD2缺失显著富集了与NF-κB转录因子活性正调节和NF-κB诱导激酶活性激活相关的基因。图3C中的GSEA数据显示NSD2丢失通过NF-κB、E2F靶点和炎症反应显著富集了与TNFα信号相关的基因。图3D显示,随着NSD2-KO细胞中p65的磷酸化增加,IκBα水平相应降低。在图3E中可以看到,KO小鼠中显示相同的结果,表明PKNf/f小鼠胰腺组织中NF-κB信号通路的激活。这些结果证明NSD2的缺失会激活NF-κB信号通路。图3F~G的TCGA数据库中患者的转录组差异分析发现,NF-κB与炎症反应和TNFα信号传导相关的基因在PDAC中显著上调,NSD2表达较低。这些发现揭示NSD2减少与NF-κB信号激活之间的联系。
4.NSD2促进Nfkbia表达并抑制p65核转位
如图4F所示,IHC发现PKNf/f小鼠胰腺组织细胞核中积累的p65蛋白比PK小鼠多。此外,在图4G中能观察到,NSD2-KO细胞系在TNF-α处理后都表现出更多的p65核转位。同时图4H中,IF分析还显示,NSD2的缺失促进了p65的核转位。这些数据表明NSD2缺失导致p65核积累增加,这进一步证明了NF-κB信号传导的激活增强。
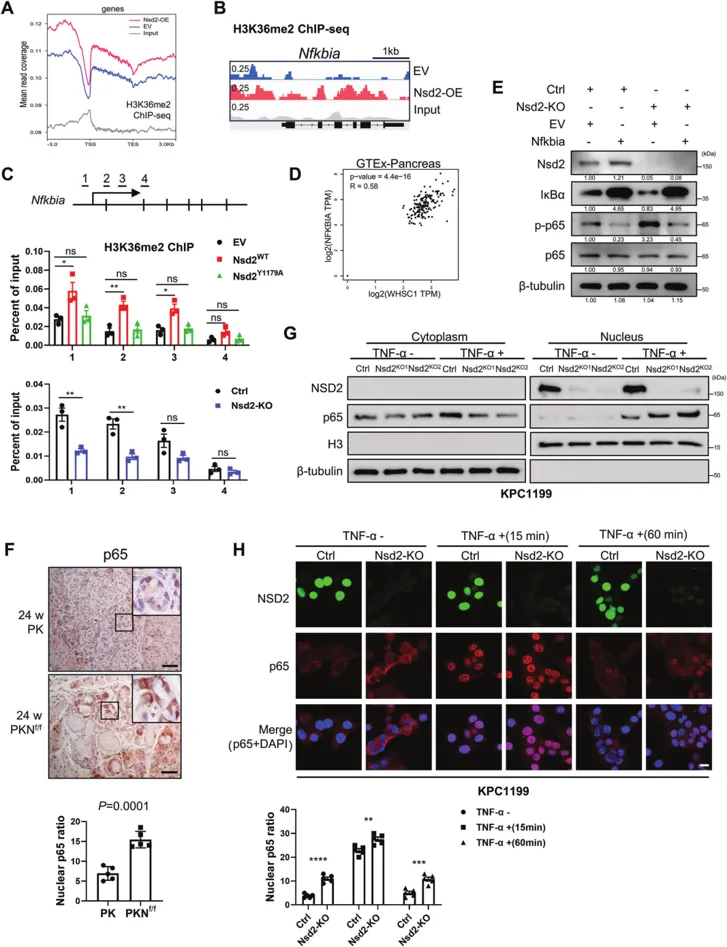
5.NSD2与p65的DNA结合域相互作用并抑制其转录活性
为了探究NSD2与p65的DNA结合域的作用关系,他们展开了相关实验,图5A显示,在TNF-α处理后15分钟,p65易位到细胞核,NSD2和p65之间的共定位发生在细胞核中。图5B中的co-IP结果显示内源性NSD2与内源性p65的相互作用。并且,图5C中的外源性co-IP还显示,NSD2和突变型NSD2Y1179A都能够与p65结合,表明NSD2和p65之间的蛋白质相互作用独立于NSD2酶活性。在图5D中,蛋白质对接发现p65通过其DNA结合结构域与NSD2相互作用。同样,图5G中EMSA显示NSD2耗竭增强了NF-κB与κB位点的结合,而NSD2过表达则使其降低。图5H~I中,ChIP-qPCR结果显示p65与这些基因的结合随着NSD2过表达而显著降低。这些结果表明NSD2与p65的DNA结合域相互作用并抑制其转录活性。
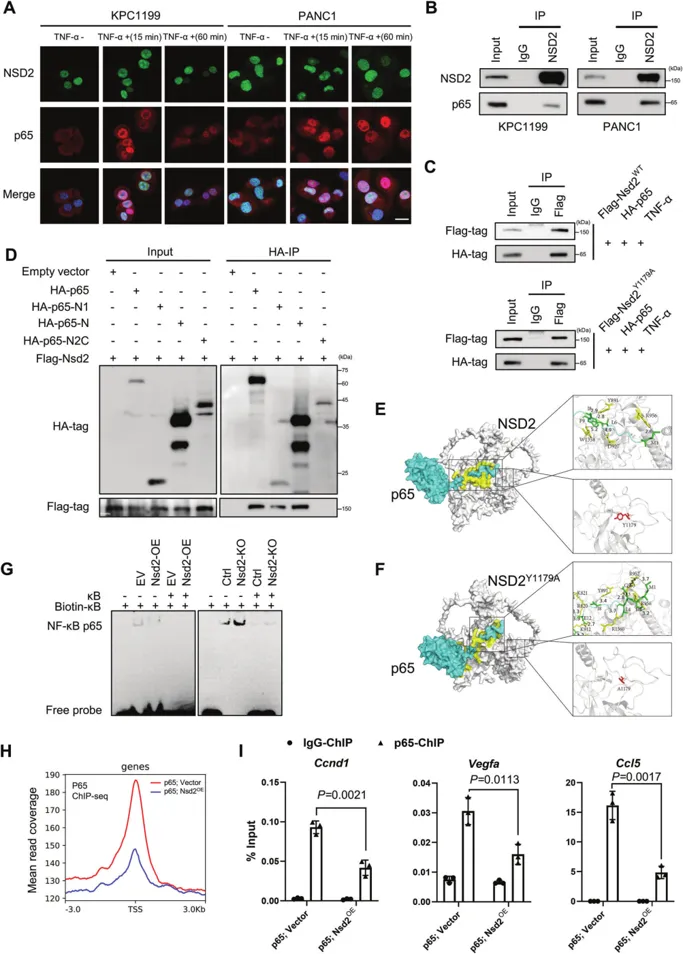
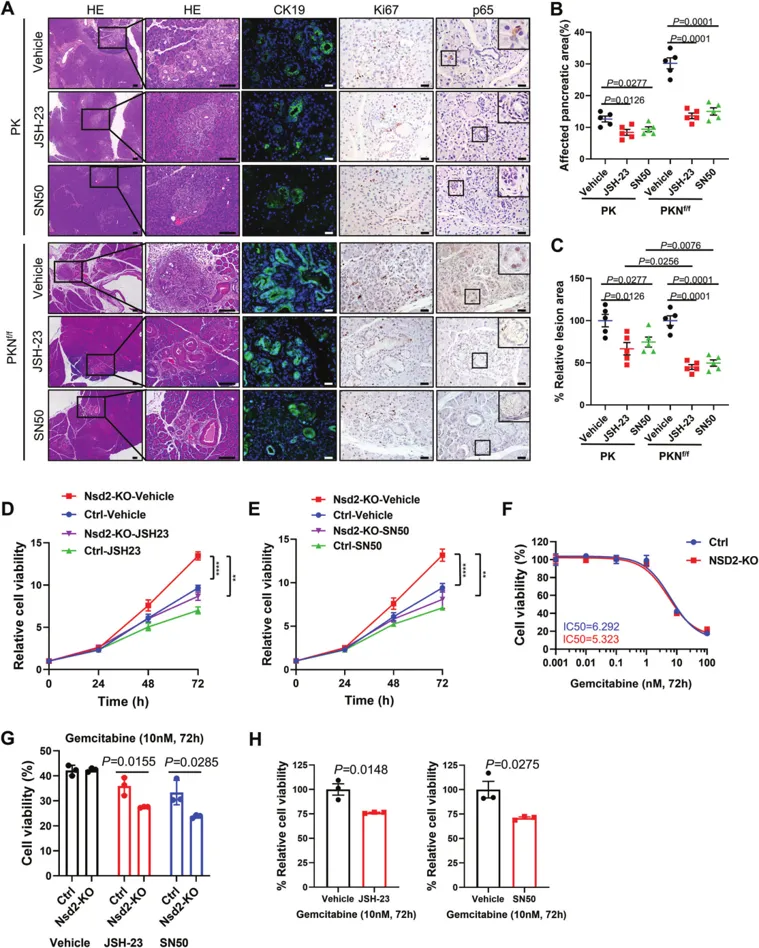
通过实验明确了NSD2与NF-κB信号通路之间的关系之后,团队开始探索其在实际胰腺病症中的应用。首先,用NF-κB抑制剂JSH-23或SN-50处理的PKNf/f小鼠显示ADM/PanIN病变和胰腺CK19和Ki67显著降低。此外,能在图6A~C中看见,小鼠都显示出p65核易位降低和p65靶基因表达水平降低。因此,抑制NF-κB信号传导缓解了小鼠NSD2缺失引起的症状。图6D~E中,JSH-23或SN50处理同样抑制了NSD2缺陷KPC1199细胞的增殖,图6G~H显示其能够使NSD2-KO细胞对适用于PDAC的药物吉西他滨更敏感。这些数据通过与吉西他滨和NF-κB抑制剂联合治疗,为NSD2低/丢失的PDAC患者提供了一种治疗策略。
五、研究小结
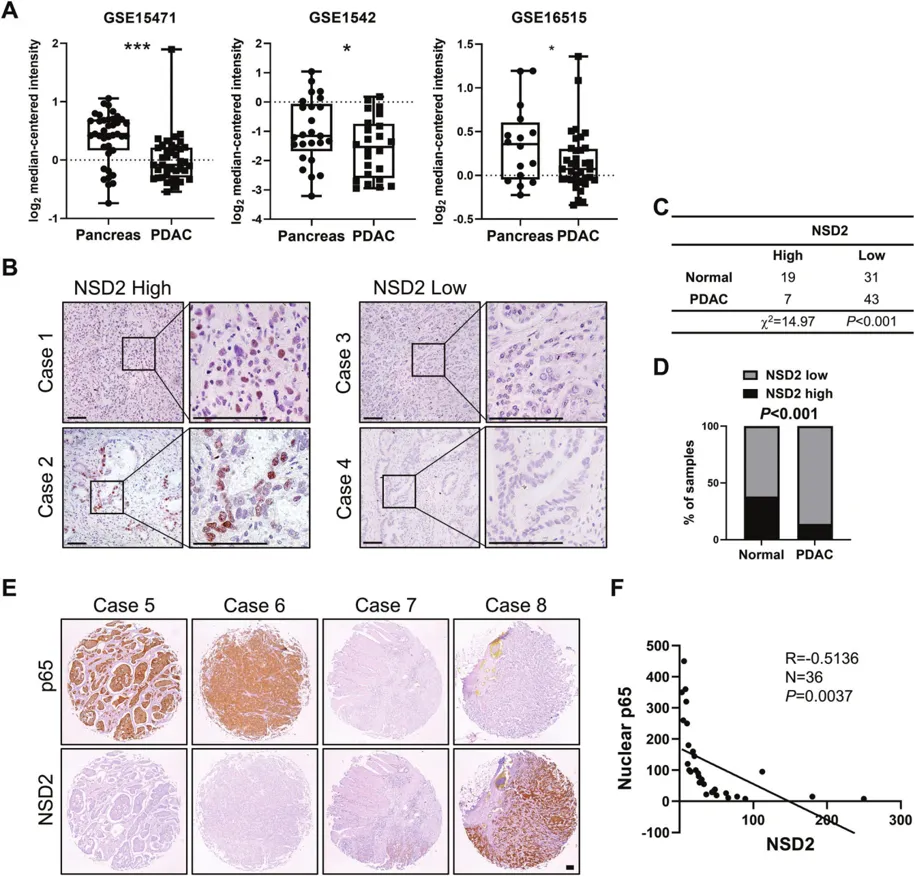
在该研究中,首先,研究人员对临床数据库进行分析,相较于正常胰腺组织,NSD2mRNA水平在PDAC患者肿瘤组织中降低,提示NSD2在胰腺癌中的功能可能与以往其它癌种报道的促癌功能不一致。接着,研究人员利用KrasG12D诱导的胰腺肿瘤小鼠模型,发现NSD2在胰腺中特异过表达可抑制小鼠的炎症和KrasG12D诱导的腺泡导管化生。反之,NSD2在胰腺特异性缺失则促进KrasG12D诱导的肿瘤发生。随后还验证了NSD2可以与p65蛋白的DNA结合域相互作用,从而直接抑制NF-κB下游转录活性。此外,利用SN50和JSH-23抑制NF-κB信号传导可缓解Nsd2缺陷PDAC小鼠的肿瘤病变表型,并使Nsd2缺陷型PDAC对吉西他滨治疗更加敏感。最后,研究人员利用临床PDAC数据库和样品,进一步证实PDAC患者的NSD2表达降低,其表达水平与核内p65水平呈负相关。
这项研究揭示了NSD2的一种新的肿瘤抑制作用机制。同时也为NSD2低表达/缺失的PDAC患者通过吉西他滨和NF-κB抑制剂联合治疗提供了新的理论思路。
六、国自然中标情况

 文章推荐
文章推荐



